
Computer System Validation (CSV) ensures pharmaceutical computer systems perform reliably and comply with regulations. Following standards like FDA 21 CFR Part 11, EU Annex 11, and PIC/S PI 011, companies can reduce validation time by 40% using GAMP 5 guidelines. However, an ISPE survey reveals 80% of pharma firms still face CSV gaps, risking compliance and product quality.
Pharma quality assurance depends on thorough CSV to maintain data integrity and regulatory adherence. Effective CSV minimizes errors, supports audits, and enhances patient safety. Continuous improvement in validation practices strengthens overall quality systems and market trust in pharmaceutical products.
What is Computer System Validation (CSV)?
Computer System Validation (CSV) confirms that computerized systems operate reliably and meet regulatory requirements. It ensures data accuracy, security, and compliance throughout the system’s lifecycle. Companies perform CSV to prevent errors and reduce risks. Also, it supports audit readiness and regulatory inspections. Proper validation enhances product quality and patient safety.
Key aspects include:
– Defining user requirements clearly
– Developing validation plans
– Executing testing protocols
– Documenting results accurately
– Performing periodic reviews and maintenance
CSV Fundamentals and Regulatory Basis
Computer System Validation (CSV) ensures that computerized systems consistently produce reliable and accurate results. It involves a documented process that verifies systems meet user requirements and comply with regulatory standards. CSV covers the entire system lifecycle, from planning and development to operation and maintenance. Proper validation reduces risks, prevents errors, and supports data integrity.
The regulatory basis for CSV includes guidelines and regulations such as:
- FDA 21 CFR Part 11 (Electronic Records and Signatures)
- EU Annex 11 (Computerized Systems in GMP)
- PIC/S PI 011 (Good Practices for Computerized Systems)
- GAMP 5 (Risk-based Approach to Validation)
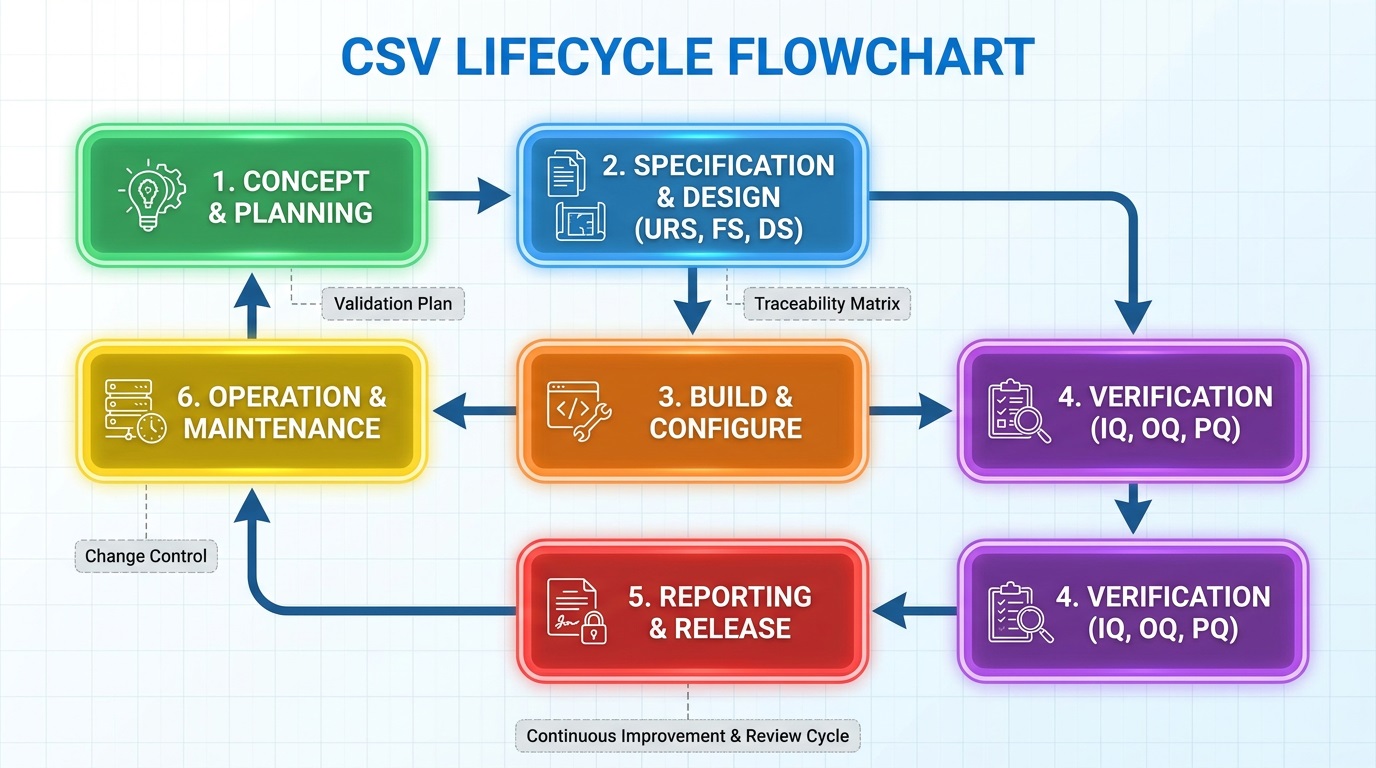
CSV Lifecycle Key Topics (Planning to Retirement)
The CSV lifecycle covers all phases from initial planning to final retirement. It ensures systems remain compliant and reliable throughout their use. Effective management reduces risks and supports consistent product quality. Also, it facilitates regulatory audits and continuous improvement.
Key lifecycle topics include:
Topic 1: Planning & Validation Master Plan (VMP
Topic 2: User/Functional Requirements (URS/FRS)
Topic 3: Testing Protocols (IQ/OQ/PQ/SAT)
Topic 4: Maintenance, Change Control & Retirement
Topic 1: Planning & Validation Master Plan (VMP)
The Validation Master Plan (VMP) outlines the overall strategy for computerized system validation. It defines scope, resources, timelines, and responsibilities. Planning a VMP ensures organized and compliant validation activities. Also, it supports regulatory readiness and risk management.
Key regulatory bases for VMP include:
FDA 21 CFR Part 11 requirements
EU GMP Annex 11 directives
PIC/S Good Practices for Computerized Systems
ICH Q7 and Q9 guidelines
GAMP 5 risk-based approach
WHO Good Manufacturing Practices
Topic 2: User/Functional Requirements (URS/FRS)
User Requirements Specification (URS) defines what users need the system to do. It focuses on high-level goals and expectations. In contrast, Functional Requirements Specification (FRS) explains how the system will fulfill these needs. FRS provides detailed descriptions of functions and features.
While URS targets stakeholders to capture user needs, FRS guides developers and testers. Together, they ensure clear communication between users and technical teams. Proper use of URS and FRS improves system design and validation success.
Topic 3: Testing Protocols (IQ/OQ/PQ/SAT)
CSV documentation includes IQ, OQ, and PQ protocols to verify installation, operation, and performance compliance. Installation Qualification (IQ) verifies that equipment is installed correctly per specifications. Operational Qualification (OQ) tests the equipment under expected operating conditions. Performance Qualification (PQ) confirms the system performs consistently in real-world scenarios. Site Acceptance Testing (SAT) checks system readiness at the installation site before full operation.
While IQ and OQ focus on equipment setup and functions, PQ evaluates performance in actual use. SAT involves the customer to ensure system meets contractual requirements. Together, these testing protocols ensure thorough validation and compliance in pharma environments.
Topic 4: Maintenance, Change Control & Retirement
Maintenance, change control, and retirement are essential for managing pharma computerized systems effectively. Maintenance keeps systems running smoothly and prevents failures. Change control manages updates to avoid unintended impacts. Retirement decommissions outdated systems responsibly, ensuring data security. Together, they sustain system integrity and compliance.
Key terms include:
- Preventive Maintenance
- Corrective Maintenance
- Change Request
- Impact Assessment
- Configuration Management
- System Decommissioning
Key Differences: CSV vs. Computer System Assurance (CSA)
Computer System Validation (CSV) focuses on documenting that systems meet intended use and regulatory requirements. It involves detailed testing and evidence collection. Conversely, Computer System Assurance (CSA) emphasizes a risk-based, flexible approach tailored to system complexity. CSA reduces unnecessary documentation while maintaining compliance.
Key differences include:
- CSV is prescriptive; CSA is risk-based.
- CSV needs exhaustive testing; CSA uses targeted verification.
- CSV requires extensive documentation; CSA recommends streamlined records.
- CSV suits highly regulated systems; CSA adapts to varied risks.
- CSV follows fixed protocols; CSA allows flexibility in approach.
- CSV timelines can be longer; CSA often reduces validation time.
Risk Management in CSV
Risk management in CSV identifies and evaluates potential system risks to prioritize validation efforts. It ensures efficient use of resources and improves system reliability. Also, it supports compliance by focusing on high-risk areas. Effective risk management reduces errors and enhances patient safety.
Key comparison points include:
- Risk identification versus risk mitigation
- Qualitative versus quantitative risk assessments
- Proactive versus reactive approaches
- Continuous monitoring versus one-time evaluation
- Focus on critical system components
- Integration with validation and quality processes
GAMP Categories 1-5 Classification
Risk management in CSV identifies and evaluates potential system risks to prioritize validation efforts. It ensures efficient use of resources and improves system reliability. Also, it supports compliance by focusing on high-risk areas. Effective risk management reduces errors and enhances patient safety.
Key comparison points include:
- Risk identification versus risk mitigation
- Qualitative versus quantitative risk assessments
- Proactive versus reactive approaches
- Continuous monitoring versus one-time evaluation
- Focus on critical system components
- Integration with validation and quality processes
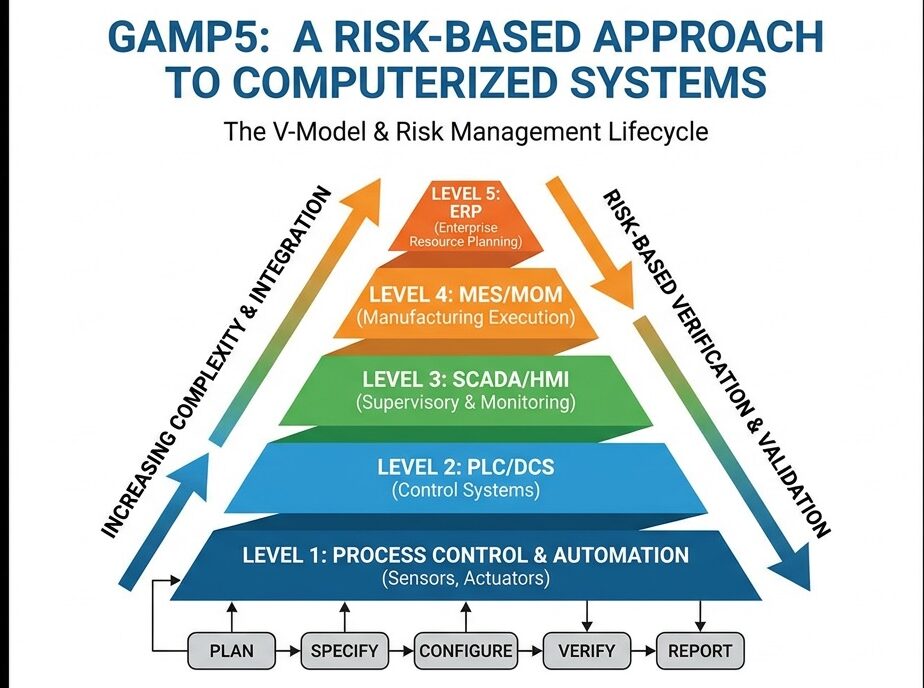
Impact and Criticality Assessment
Impact and criticality assessment evaluates how system failures affect product quality and patient safety. It helps prioritize validation efforts by focusing on high-risk systems first. Accurate assessment prevents resource waste and reduces compliance risks. Also, it supports regulatory expectations for risk-based validation.
This process improves decision-making throughout the CSV lifecycle. It identifies critical controls needing strict monitoring and maintenance. Furthermore, impact assessment guides corrective actions and continuous improvement. Overall, it strengthens pharma quality assurance and regulatory compliance.
CSV Checklists & Protocols
CSV checklists and protocols guide systematic validation of computerized systems in pharma. They ensure all validation steps are completed accurately and consistently. Moreover, well-defined protocols help avoid errors and meet regulatory requirements. Proper use increases audit readiness and improves quality control.
Key items include:
- Validation Master Plan (VMP)
- User Requirements Specification (URS)
- Installation Qualification (IQ), Operational Qualification (OQ), Performance Qualification (PQ)
- Change Control Procedures
| Computer System Validation (CSV) Protocol Checklist | Description |
|---|---|
|
Validation Master Plan (VMP) prepared and approved |
Defines scope, responsibilities, timeline |
|
User Requirements Specification (URS) documented |
Clearly states user needs and expectations |
|
Functional Requirements Specification (FRS) created |
Details system functions and workflows |
|
Risk Assessment completed |
Identifies potential risks and mitigations |
|
Qualification (IQ- OQ-PQ) protocol executed |
Verifies proper installation of equipment |
Final Words
CSV ensures GxP trust by validating computerized systems following industry best practices. Implementing GAMP 5 helps pharma companies reduce validation time by up to 40%, ensuring compliance with ICH guidelines like Q7 and Q9. For effective audits, consult FDA 21 CFR Part 11 regulations and download comprehensive checklists tailored for CSV.
Key pharma references include FDA FY23 OAI statistics (fda.gov) and ISPE GAMP 5 case studies. These resources highlight common compliance challenges and solutions. Share your CSV challenges below—what’s your biggest hurdle in ensuring regulatory adherence?
FAQs
Computer System Validation, or CSV, is the documented process of proving that a computerized system works as intended. In pharma, CSV helps protect data integrity, product quality, GMP compliance, and patient safety.
CSV helps QA teams confirm that digital systems produce accurate, secure, and reliable records. It also supports audit readiness, reduces compliance risks, and helps companies meet regulatory expectations.
CSV focuses on documented validation evidence. CSA, or Computer System Assurance, uses a more risk-based approach. It focuses testing effort on high-risk functions while reducing unnecessary documentation for low-risk areas.
References

Ershad Moradi
Ershad Moradi, a Content Marketing Specialist at Zamann Pharma Support, brings 6 years of experience in the pharmaceutical industry. Specializing in pharmaceutical and medical technologies, Ershad is currently focused on expanding his knowledge in marketing and improving communication in the field. Outside of work, Ershad enjoys reading and attending industry related networks to stay up-to-date on the latest advancements. With a passion for continuous learning and growth, Ershad is always looking for new opportunities to enhance his skills and contribute to pharmaceutical industry. Connect with Ershad on Facebook for more information.

Pharma Regulatory Consulting Firms 2026: Unlock Your Drug Approval Success
Pharma regulatory consulting firms provide critical expertise that accelerates drug approvals and ensures global compliance. By navigating complex regulations like FDA, EMA, and ICH, these firms reduce risks and optimize submission quality. This blog explores the consulting role, benefits, and key regulations, encouraging knowledge sharing to enhance industry success.

QMS in Pharmaceutical Manufacturing 2026: Unlock Quality & Compliance Success
A robust Quality Management System (QMS) forms the backbone of pharmaceutical manufacturing. It ensures adherence to GMP and global standards, mitigates risks, controls processes, and drives continuous improvement. By maintaining accurate documentation and training staff, QMS helps pharma companies achieve regulatory compliance and deliver safe, high-quality products consistently.

Pharma QMS Interview Questions and Answers: GMP and QA Guide in 2026
This article explains pharma QMS interview questions and answers with a focus on GMP compliance interview questions, CAPA scenarios, deviation handling, and FDA inspection readiness QA. It helps candidates prepare for real pharmaceutical quality assurance interview expectations.