
Medical device regulatory submissions are essential to gain market approval and ensure patient safety. According to WHO, nearly 30% of medical device approvals involve 510(k) submissions, which demonstrate substantial equivalence. Premarket Approval (PMA) is required for higher-risk devices, while CE marking indicates compliance with EU regulations. These submissions demand thorough documentation, robust risk assessments, and strong clinical evidence to support safety and effectiveness.
Pharma regulation increasingly emphasizes transparency and data integrity in medical device approvals. Companies must align with global requirements, maintaining detailed records and validating risk controls. This comprehensive approach reduces regulatory delays and enhances product trust. Therefore, understanding diverse submission types is vital for successful pharma regulation compliance and timely market access.
Table of Contents
What is Medical Device Regulatory Submissions?
Medical device regulatory submissions allow manufacturers to get approval for market entry. They include key types like 510(k), PMA, and CE marking. According to FDA, 70% of device approvals use 510(k) submissions. Submissions require evidence of safety, performance, and manufacturing quality. These steps protect patients and ensure regulatory compliance.
Submissions also involve:
- Detailed device description
- Risk assessments and mitigation plans
- Clinical data supporting effectiveness
- Manufacturing and quality system documentation
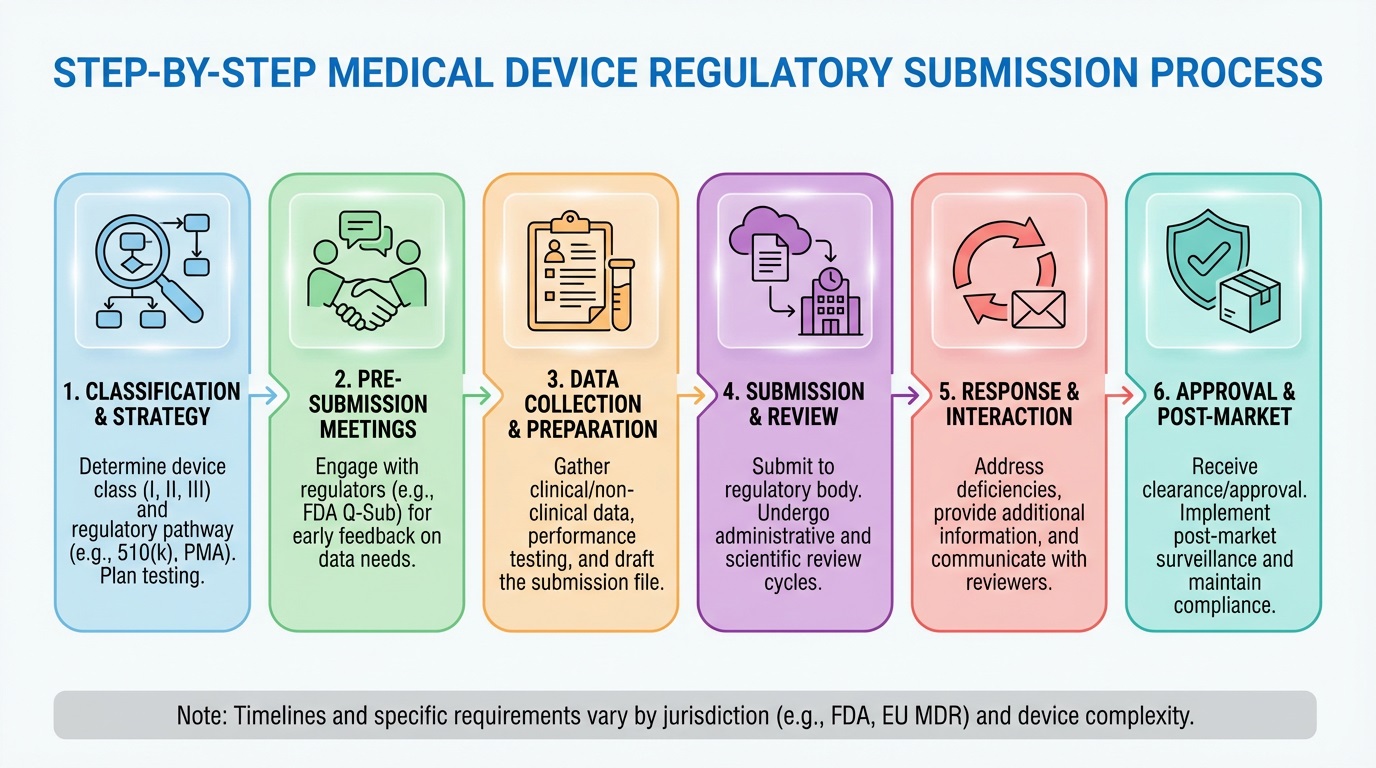
Step-by-step Process for Preparing Regulatory Submissions for Medical Devices
To prepare regulatory submissions for medical devices, start by gathering all required documents. Include device description, design details, and testing data. Then, conduct thorough risk assessments and clinical evaluations to prove safety and effectiveness. Next, compile and format all files according to FDA or EU MDR guidelines. Finally, submit the application through the correct portal and respond quickly to regulatory inquiries.
Key steps include:
- Step 1: Gather and organize required documentation
- Step 2: Conduct risk assessments and clinical evaluations
- Step 3: Compile and format submission files
- Step 4: Submit the application
Step 1: Gather and Organize Required Documentation
Start preparing regulatory submissions by gathering all necessary documentation carefully. Collect detailed device descriptions, design specifications, and testing data. Also, organize supplier information and manufacturing processes clearly. This step builds a strong foundation for successful submission. Proper documentation supports regulatory review and speeds approval.
Ensure documents are complete, accurate, and up to date. Collaborate with cross-functional teams to verify data consistency.
Key tasks include:
- Collect device description and specifications
- Compile testing and validation data
- Organize supplier and manufacturing details
- Maintain document version control
Step 2: Conduct Risk Assessments and Clinical Evaluations
During Step 2, teams conduct risk assessments to identify potential hazards and control measures. They also perform clinical evaluations to prove device safety and effectiveness. Moreover, they analyze data to support regulatory requirements. Clear risk management helps prevent failures and protects patients.
Key questions include:
– What risks does the device pose?
– How effective is the device clinically?
– When should evaluations be updated?
– Who reviews and approves risk assessments?
Step 3: Compile and Format Submission Files
In Step 3, compile submission files carefully following FDA or EU MDR guidelines. Ensure all documents are complete and well-organized. Also, format files according to regulatory requirements and templates. This improves clarity and accelerates review. Accurate submissions reduce questions and delays.
Key steps include:
- Gather all required documents
- Organize files by submission sections
- Follow specific formatting guidelines
- Include all certificates and approvals
- Check for data consistency
- Review before final submission
Step 4: Submit the Application
In Step 4, submit the application through the correct regulatory portal, such as FDA’s or EU MDR’s online system. Verify all required documents are uploaded properly. Also, monitor submission status regularly and address any agency questions quickly. Prompt responses help avoid delays and improve approval chances.
Key actions include:
- Confirm submission portal and format
- Upload all files completely
- Verify submission receipt confirmation
- Monitor progress actively
- Respond promptly to agency inquiries
Key Documents Required for Regulatory Submissions
Key documents in medical device regulatory submissions vary by region and risk level. Premarket Notifications (510(k)) demonstrate substantial equivalence to legally marketed devices. PMA submissions provide scientific evidence for high-risk devices. Also, CE Marking shows compliance with European Union regulations. Properly preparing these documents speeds review and approval.
Important documents include:
– Premarket Notifications (510(k))
– PMA
– CE Marking
Checklist of Documents for 510(k), PMA, and CE Marking
| Document Type | Key Required Documents | Purpose |
|---|---|---|
|
510(k) Premarket Notification |
Device description, substantial equivalence, test reports |
Demonstrate equivalence to existing device |
|
PMA (Premarket Approval) |
Clinical data, manufacturing details, risk analysis |
Prove safety and effectiveness for high-risk devices |
|
CE Marking |
Technical file, risk management, clinical evaluation |
Show compliance with EU MDR requirements |
Premarket Notifications (510(k))
Premarket Notifications (510(k)) require several key documents to ensure regulatory compliance. Submitters must provide device descriptions and substantial equivalence comparisons. Also, include test reports and labeling information. Clear documentation supports FDA review and answers inquiries efficiently.
Key documents include:
- Device description and intended use
- Substantial equivalence comparison
- Performance and safety test reports
- Labeling and instructions for use
- Software validation documents (if applicable)
- Risk management and mitigation plans
Premarket Approval (PMA)
Premarket Approval (PMA) requires comprehensive documentation to prove device safety and effectiveness. Companies must submit detailed clinical data and manufacturing processes. Also, risk assessments help identify potential hazards. Clear, organized documents support regulatory review and approval.
Key PMA documents include:
- Clinical study reports
- Device description and design details
- Manufacturing process documentation
- Risk analysis and mitigation plans
- Labeling and instructions for use
Quality system compliance evidence
CE Marking
CE Marking requires a technical file demonstrating compliance with EU regulations. Companies prepare detailed risk assessments and clinical evaluations. Also, they develop instructions for use and labeling. Organized documentation helps regulatory bodies approve the device efficiently.
Key CE Marking documents include:
- Technical documentation
- Risk management files
- Clinical evaluation report
- Labeling and user manuals
- Declaration of conformity
- Quality management system certificates
Technical Files and Design Dossiers for Medical Device Submissions
Technical files and design dossiers contain detailed information about medical device design and safety. They include data on materials, manufacturing, and performance testing. Also, they provide proof of compliance with regulations. Proper preparation ensures faster review and approval.
For example:
- A hip implant’s design dossier details biocompatibility and mechanical testing.
- A diagnostic device’s technical file includes software validation documents.
- An inhaler’s file covers production methods and risk assessments.
These examples show the importance of complete, accurate documentation.
Understanding EU MDR and IVDR Requirements
The EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) set strict rules. They aim to improve device safety and performance across Europe. Manufacturers must comply with new clinical evaluation and post-market surveillance requirements. These regulations enhance transparency and traceability for all medical devices.
Additionally, both MDR and IVDR require thorough technical documentation and risk management plans. Also, companies must implement quality management systems aligned with EU standards. This ensures continuous monitoring and reporting of device safety. Compliance reduces regulatory risks and improves patient confidence.
Differences Between Global Medical Device Regulatory Frameworks
Global medical device regulatory frameworks include FDA (USA), EU MDR (Europe), PMDA (Japan), and Health Canada. These frameworks differ in approval processes, documentation requirements, and risk classifications. Understanding these differences helps companies tailor submissions for each market effectively.
Moreover, frameworks vary in clinical data demands and post-market surveillance rules. Also, timelines and fees differ, impacting product launch speed. Companies must align their strategies with regional regulations to ensure compliance and market access across borders.

Final words
Medical device regulatory submissions are critical for gaining timely approvals and achieving market success. Studies show that well-prepared submissions can reduce review times by up to 35%, improving time-to-market. Comprehensive documentation, clinical evidence, and risk management increase regulator trust. Manufacturers following thorough submission protocols face fewer delays and recall risks, ensuring patient safety and business growth.
Moreover, continuous education and adherence to evolving regulations remain essential. Industry data indicates that ongoing training reduces compliance errors by 25%. Staying updated with global guidelines helps companies maintain product quality and avoid penalties. Therefore, commitment to regulatory knowledge supports sustainable success.
FAQs
Medical device regulatory submissions are formal requests to obtain market approval, ensuring device safety and effectiveness.
The main types include Premarket Notifications (510(k)), Premarket Approval (PMA), and CE Marking for the EU.
Submissions require device descriptions, risk assessments, clinical data, manufacturing documentation, and labeling information.
References

Ershad Moradi
Ershad Moradi, a Content Marketing Specialist at Zamann Pharma Support, brings 6 years of experience in the pharmaceutical industry. Specializing in pharmaceutical and medical technologies, Ershad is currently focused on expanding his knowledge in marketing and improving communication in the field. Outside of work, Ershad enjoys reading and attending industry related networks to stay up-to-date on the latest advancements. With a passion for continuous learning and growth, Ershad is always looking for new opportunities to enhance his skills and contribute to pharmaceutical industry. Connect with Ershad on Facebook for more information.

Pharma Regulatory Consulting Firms 2026: Unlock Your Drug Approval Success
Pharma regulatory consulting firms provide critical expertise that accelerates drug approvals and ensures global compliance. By navigating complex regulations like FDA, EMA, and ICH, these firms reduce risks and optimize submission quality. This blog explores the consulting role, benefits, and key regulations, encouraging knowledge sharing to enhance industry success.

QMS in Pharmaceutical Manufacturing 2026: Unlock Quality & Compliance Success
A robust Quality Management System (QMS) forms the backbone of pharmaceutical manufacturing. It ensures adherence to GMP and global standards, mitigates risks, controls processes, and drives continuous improvement. By maintaining accurate documentation and training staff, QMS helps pharma companies achieve regulatory compliance and deliver safe, high-quality products consistently.

Pharma QMS Interview Questions and Answers: GMP and QA Guide in 2026
This article explains pharma QMS interview questions and answers with a focus on GMP compliance interview questions, CAPA scenarios, deviation handling, and FDA inspection readiness QA. It helps candidates prepare for real pharmaceutical quality assurance interview expectations.