
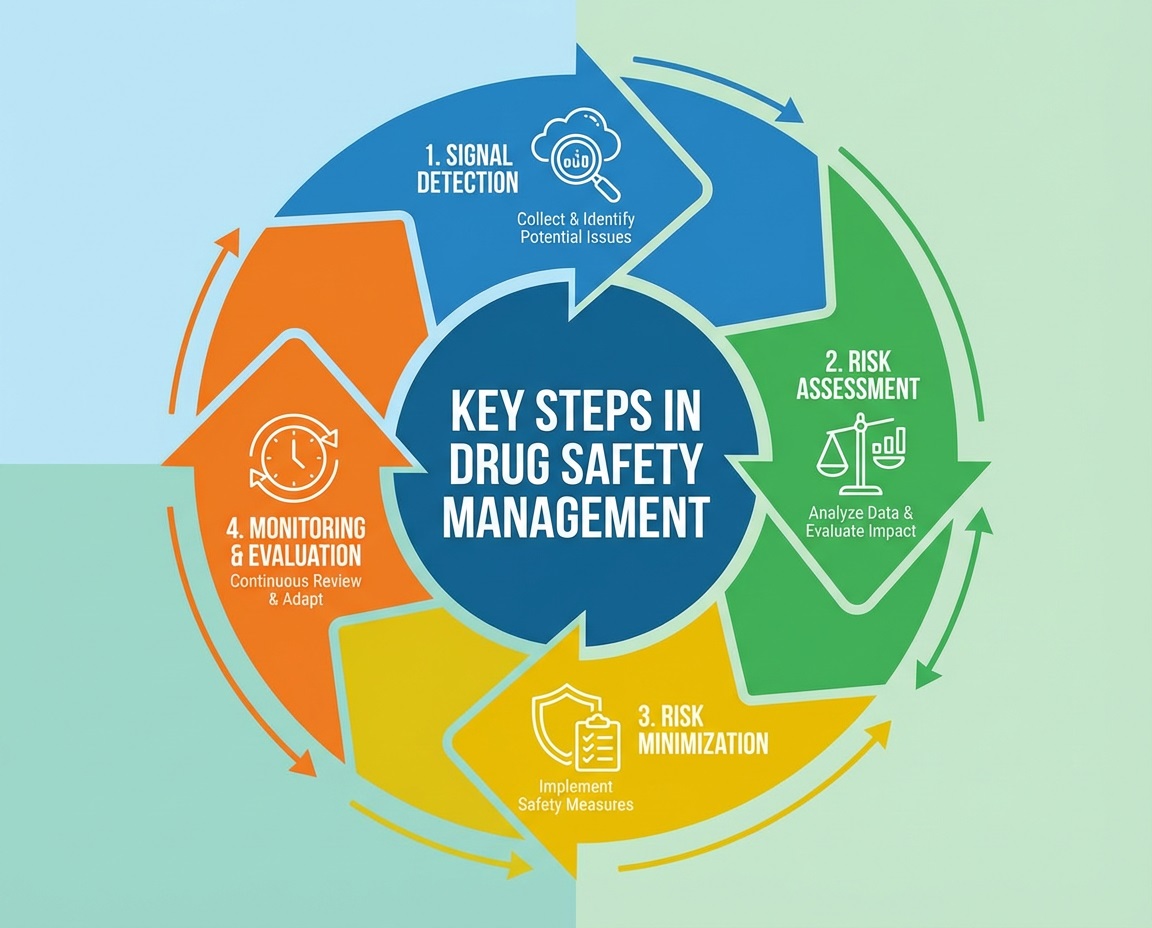
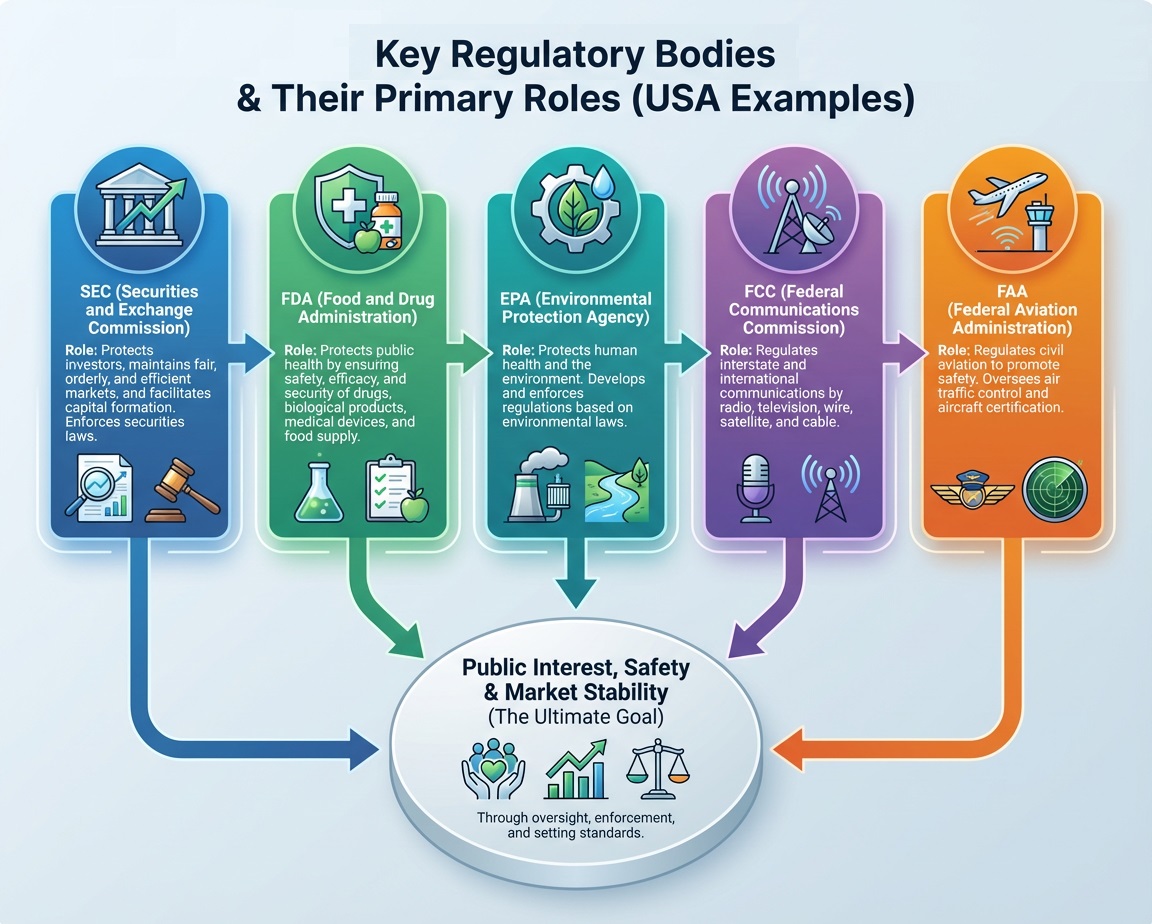


Pharma Regulatory Consulting Firms 2026: Unlock Your Drug Approval Success
Pharma regulatory consulting firms provide critical expertise that accelerates drug approvals and ensures global compliance. By navigating complex regulations like FDA, EMA, and ICH, these firms reduce risks and optimize submission quality. This blog explores the consulting role, benefits, and key regulations, encouraging knowledge sharing to enhance industry success.

QMS in Pharmaceutical Manufacturing 2026: Unlock Quality & Compliance Success
A robust Quality Management System (QMS) forms the backbone of pharmaceutical manufacturing. It ensures adherence to GMP and global standards, mitigates risks, controls processes, and drives continuous improvement. By maintaining accurate documentation and training staff, QMS helps pharma companies achieve regulatory compliance and deliver safe, high-quality products consistently.

Pharma QMS Interview Questions and Answers: GMP and QA Guide in 2026
This article explains pharma QMS interview questions and answers with a focus on GMP compliance interview questions, CAPA scenarios, deviation handling, and FDA inspection readiness QA. It helps candidates prepare for real pharmaceutical quality assurance interview expectations.