


Drug Regulatory Agencies Complete Guide 2026: Regulatory Approval & Compliance
This blog explores the vital roles of Drug Regulatory Agencies like the FDA and the EMA Regulatory Standards in ensuring the safety and efficacy of medicines globally. By reviewing thousands of applications each year, these agencies protect public health and facilitate access to life-saving drugs.

Pharma Jobs in Houston in 2026: FDA GMP Manufacturing and Salary Outlook
Houston’s life sciences sector continues to expand across pharmaceutical jobs in Houston, biotech manufacturing jobs Houston, and GMP jobs in Houston, driven by FDA inspections Texas activity, sterile manufacturing compliance standards, and growing clinical research infrastructure.

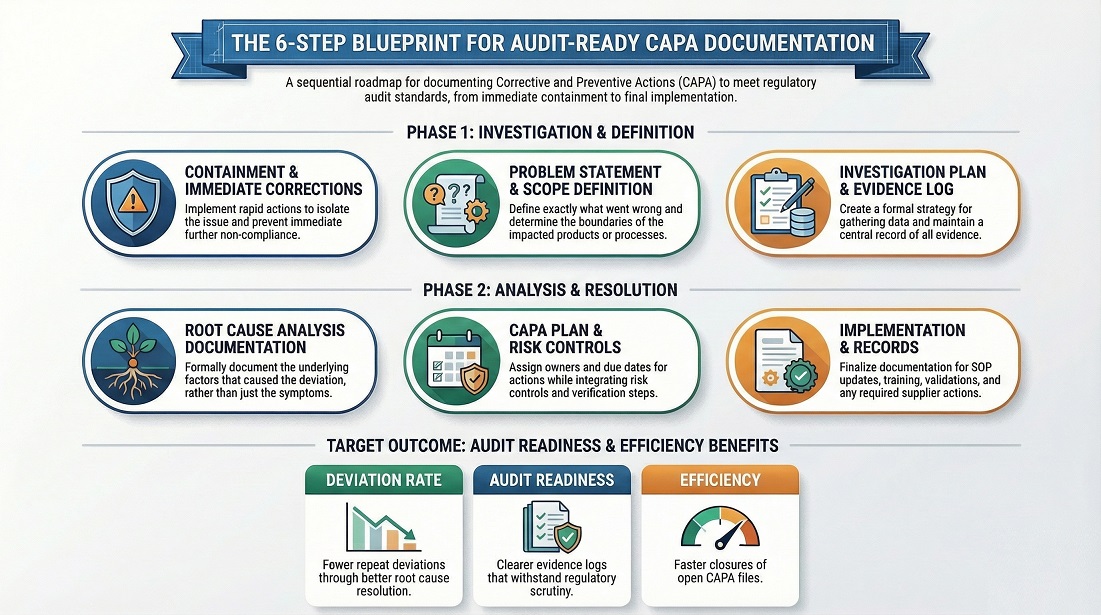
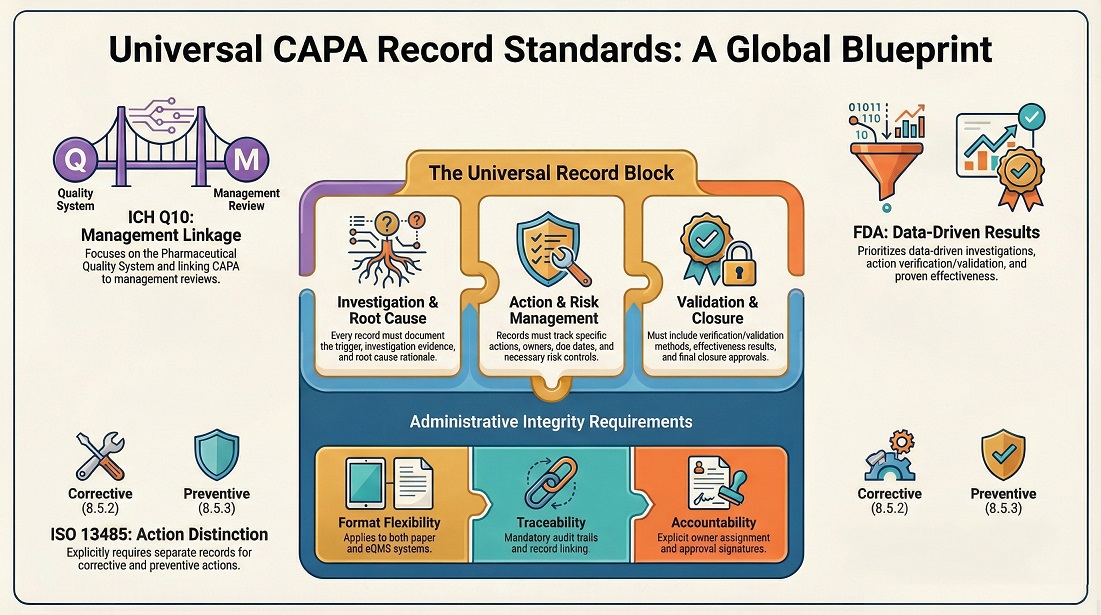
Key Performance Indicator (KPI) Guide for Pharmaceutical Industry 2026 : Complete Quality Metrics
Approximately 30% of FDA Warning Letters cite inadequate monitoring of Key Performance Indicators (KPIs) as a major compliance issue, highlighting their crucial role in pharmaceutical operations. KPIs provide measurable data to track process efficiency, product quality, and regulatory adherence. Proper KPI management supports risk mitigation, enhances decision-making, and ensures patient safety.