
In recent GMP inspection cycles, regulators have linked 25–35% of repeat observations directly to Process Deviations, making them one of the most persistent inspection drivers across pharmaceutical manufacturing sites. Inspectors no longer view deviations as isolated events; instead, they treat them as regulatory evidence of how well a site’s quality system performs under real operating pressure. For QA teams, deviation handling now functions as a live audit of control maturity, documentation discipline, and decision logic across the entire operation. This is why deviation records increasingly shape inspection outcomes as much as validation or data integrity.
Within modern inspection models, deviation files also connect directly to broader expectations in pharma quality assurance, where regulators assess system behavior rather than isolated compliance actions.
Table of Contents
What Process Deviations Mean in a GMP Inspection Context
Process deviations represent formal GMP events that signal potential loss of control, not routine execution errors.
A process deviation refers to any unplanned departure from an approved manufacturing or quality procedure that may affect product quality, patient safety, or regulatory compliance.
Inspectors define deviations based on regulatory impact, not internal convenience. A missed in-process check, an undocumented parameter adjustment, or delayed environmental monitoring review can all qualify if they undermine procedural intent. During inspections, regulators scope deviations across batches, shifts, products, and timeframes to detect patterns. They do not accept narrow event framing when broader control questions remain unanswered. As a result, deviations become indicators of how consistently procedures operate under normal production conditions.
Why Deviation Management Weaknesses Lead to Inspection Findings
The following flow visualizes how a single process deviation can escalate through investigation gaps and CAPA weaknesses into a formal inspection finding.
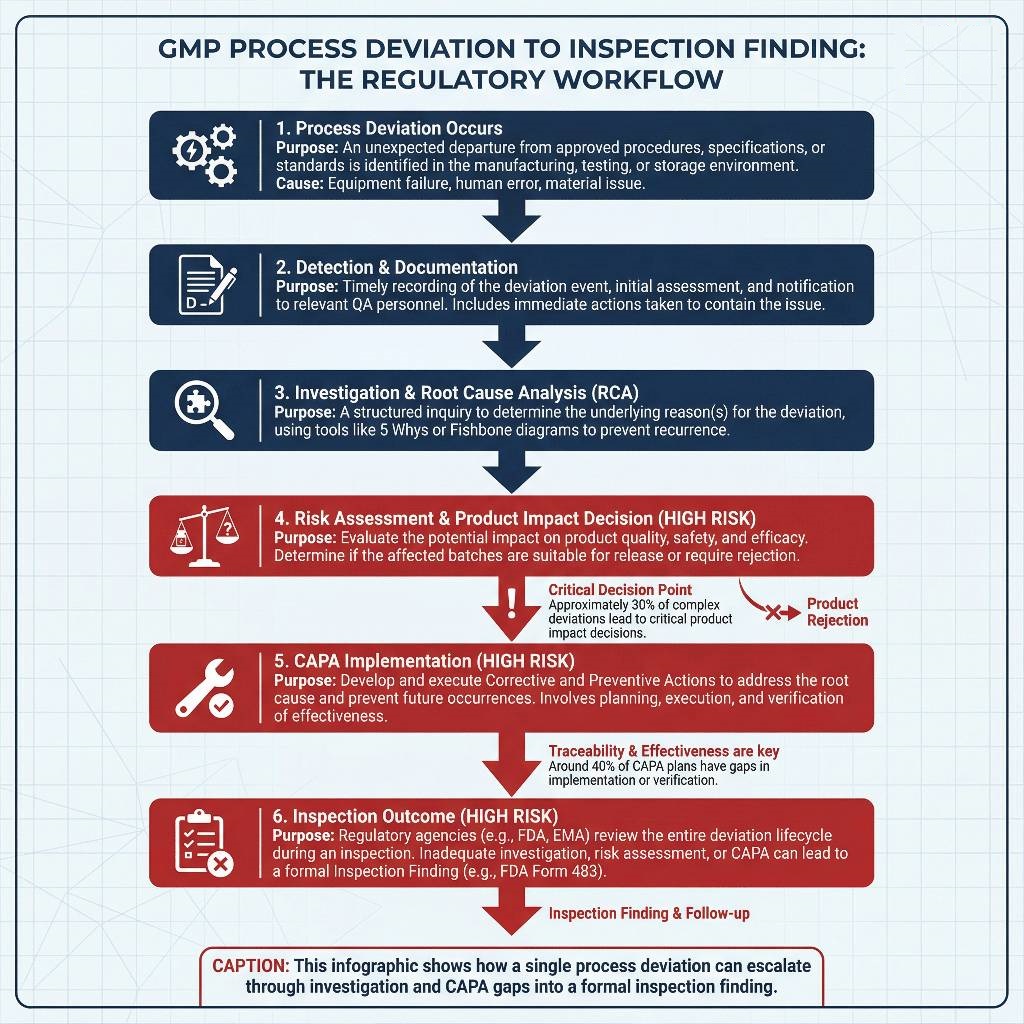
Weak deviation systems trigger findings because they expose systemic quality risk rather than isolated mistakes.
Delayed investigations, shallow root cause analysis, and inconsistent documentation suggest that quality decisions occur reactively. Inspectors interpret these signals as governance gaps. When similar manufacturing deviations receive different classifications or CAPA actions, regulators infer subjective decision-making. Over time, this inconsistency drives repeat observations because unresolved root causes continue to surface across inspection cycles.
How Inspectors Evaluate Deviation Handling Within Pharmaceutical Quality Systems
Deviation handling is assessed as an integrated function of the quality system rather than a standalone workflow. Effective evaluation focuses on consistency in decisions, logical documentation, and evidence that actions are reliable under normal production conditions. Well-managed deviations demonstrate that the site applies standardized processes across similar events and maintains traceability from detection to closure.
The core inspection focus areas that shape deviation assessment include:
- Detection and documentation of manufacturing deviations
- Deviation investigation and root cause analysis logic
- Deviation risk assessment and product impact decisions
- CAPA effectiveness and systemic prevention of recurrence
Detection and Documentation of Manufacturing Deviations
Deviation handling is assessed as an integrated function of the quality system rather than a standalone workflow. Effective evaluation focuses on consistency in decisions, logical documentation, and evidence that actions are reliable under normal production conditions. Well-managed deviations demonstrate that the site applies standardized processes across similar events and maintains traceability from detection to closure.
The core inspection focus areas that shape deviation assessment include:
- Detection and documentation of manufacturing deviations
- Deviation investigation and root cause analysis logic
- Deviation risk assessment and product impact decisions
- CAPA effectiveness and systemic prevention of recurrence
Detection and Documentation of Manufacturing Deviations
Immediate detection and real-time documentation of deviations are critical for regulatory compliance and data integrity. Delays between event occurrence and documentation raise data integrity concerns. Records must clearly link the deviation event, batch context, and initial impact assessment. Ambiguous narratives or retrospective corrections undermine credibility. In practice, inspectors often compare logbooks, batch records, and deviation files line by line to confirm alignment under operational conditions.
Deviation Investigation and Root Cause Analysis Logic
Investigations that stop at human error or procedural noncompliance often face regulatory scrutiny. They expect evidence-based root cause analysis supported by data trends, equipment history, and process variability. Unsupported assumptions fail inspection scrutiny. During inspections, regulators frequently request raw data to confirm whether investigation conclusions reflect actual process behavior.
Deviation Risk Assessment and Product Impact Decisions
Deviation risk classification must align with scientific rationale. Inspectors assess whether severity ratings match potential product impact and patient exposure. They also verify consistency across similar deviations. When low-risk classifications accompany extensive investigations, or high-risk deviations receive limited follow-up, regulators identify decision logic breakdowns.
CAPA Effectiveness and Systemic Prevention of Recurrence
The next visual highlights the ideal logic for conducting deviation investigations and ensuring CAPA effectiveness, emphasizing measurable outcomes and systemic prevention of recurrence.
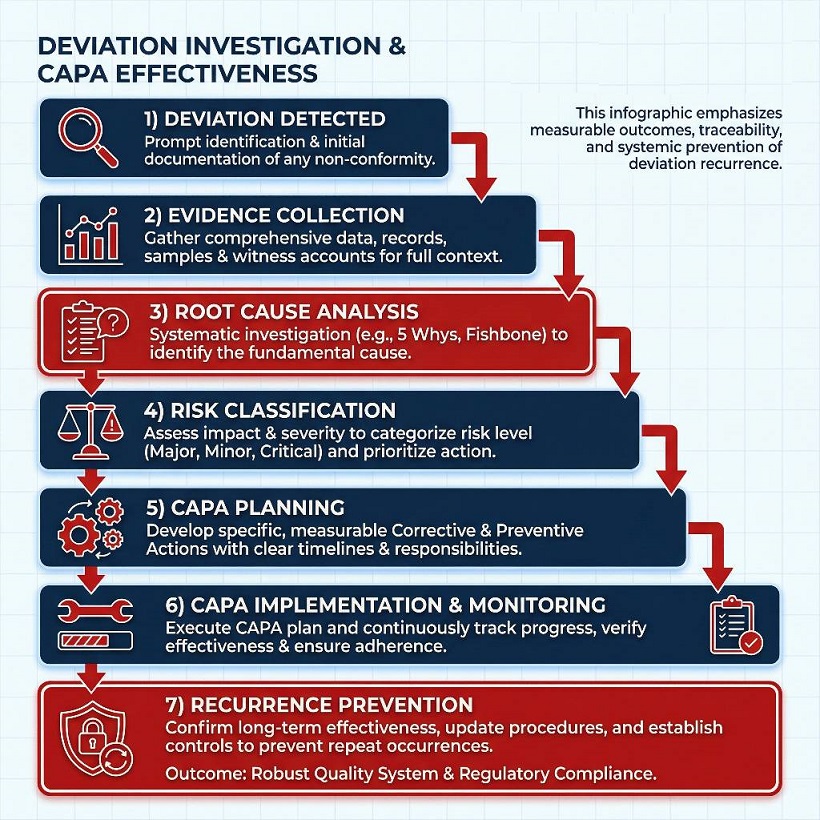
CAPA success is evaluated based on its ability to prevent recurrence rather than simply meeting closure deadlines. Effective CAPAs show measurable outcomes, defined monitoring plans, and integration into change control or training systems. In one inspection scenario, a site closed CAPAs on time but still received findings because similar deviations reappeared, indicating ineffective systemic correction.
Common Deviation Management Gaps Identified During Inspections
Recurrent findings consistently reveal similar deviation control weaknesses across multiple sites:
| Deviation management gap | What inspectors look for | Why it triggers findings |
|---|---|---|
|
Late deviation initiation |
Timely detection and documentation |
Signals delayed quality oversight |
|
Weak root cause analysis |
Evidence-based causal logic |
Indicates unresolved systemic risk |
|
Inconsistent risk classification |
Alignment across similar deviations |
Suggests subjective decision-making |
|
Ineffective CAPAs |
Measurable prevention of recurrence |
Leads to repeat observations |
|
Missing deviation trending |
Use of trend data in management review |
Shows lack of quality governance |
These gaps signal to inspectors that Process Deviations remain operationally tolerated rather than actively controlled, increasing the likelihood of repeat GMP observations.
How Effective Deviation Management Supports Inspection Readiness
Structured deviation management strengthens inspection readiness by demonstrating control maturity and consistent decision-making. A well-documented deviation system provides clear, traceable evidence that quality decisions are applied consistently across all routine manufacturing operations.
When deviation handling follows logical procedures, regulators spend less time questioning outcomes and more time verifying overall system robustness. Consistent classification, justified risk assessments, and effective CAPAs minimize follow-up inquiries. Across multiple inspection cycles, this stability reinforces regulatory confidence and reduces the likelihood of repeat findings linked to manufacturing deviations GMP.
Final Word
In recent GMP inspection programs, deviation-related deficiencies account for roughly 30% of repeat observations across manufacturing sites. In one notable case, a mid-size sterile injectable facility recorded over 200 deviations in a single year, yet only 20% were escalated to high-risk classification, and fewer than half resulted in CAPAs with measurable outcomes. Inspectors did not challenge the volume of deviations; they focused on whether the risk assessments were scientifically justified, investigations were thorough, and CAPAs truly prevented recurrence.
This pattern illustrates why Process Deviations are now considered concrete regulatory evidence of system control. Sites that allow repeated low-risk deviations without proper follow-up signal weak governance, whereas those aligning investigation depth, risk logic, and CAPA effectiveness significantly reduce inspection pressure and stabilize audit timelines.
FAQs
Inspectors prioritize deviations that affect batch release decisions, critical process parameters, and manufacturing controls. Repeated deviations of the same type and undocumented process changes also raise concerns about sustained loss of control within the quality system.
Because many investigations rely on assumptions instead of objective evidence, such as deviation trends, equipment history, or process data. When the investigation does not explain why existing controls failed, inspectors consider the root cause unsupported.
Consistent risk classification logic, CAPAs linked to measurable outcomes, and trending deviations across products and time. Inspectors gain confidence when deviation data clearly feeds into management review and system-level prevention.
References

Mahtab Shardi
Mahtab is a pharmaceutical professional with a Master’s degree in Physical Chemistry and over five years of experience in laboratory and QC roles. Mahtab contributes reliable, well-structured pharmaceutical content to Pharmuni, helping turn complex scientific topics into clear, practical insights for industry professionals and students.

Pharma Regulatory Consulting Firms 2026: Unlock Your Drug Approval Success
Pharma regulatory consulting firms provide critical expertise that accelerates drug approvals and ensures global compliance. By navigating complex regulations like FDA, EMA, and ICH, these firms reduce risks and optimize submission quality. This blog explores the consulting role, benefits, and key regulations, encouraging knowledge sharing to enhance industry success.

QMS in Pharmaceutical Manufacturing 2026: Unlock Quality & Compliance Success
A robust Quality Management System (QMS) forms the backbone of pharmaceutical manufacturing. It ensures adherence to GMP and global standards, mitigates risks, controls processes, and drives continuous improvement. By maintaining accurate documentation and training staff, QMS helps pharma companies achieve regulatory compliance and deliver safe, high-quality products consistently.

Pharma QMS Interview Questions and Answers: GMP and QA Guide in 2026
This article explains pharma QMS interview questions and answers with a focus on GMP compliance interview questions, CAPA scenarios, deviation handling, and FDA inspection readiness QA. It helps candidates prepare for real pharmaceutical quality assurance interview expectations.